
Types of sequencing for genomic or metagenomic DNA
Illumina sequencing
Illumina sequencing is a widely used and accurate method for high-throughput DNA sequencing. This method uses Illumina’s sequencing by synthesis (SBS) technology, where fluorescently-tagged nucleotides are optically tracked during DNA synthesis. The sequence reads are typically 150 base pairs in length in both the forward and reverse directions. We use Illumina’s DNA prep kit which is flexible for genomes of all sizes. Using unique dual indexes (also called barcodes), we can also multiplex samples on the same flowcell, enabling economies of scale which provides you with cost savings.
Oxford Nanopore Technologies sequencing
Auckland Genomics staff are experts in Oxford Nanopore Technologies sequencing. Oxford Nanopore Technologies (ONT) sequencing is designed to sequence genetic material of any length, with the potential to sequence the full length of a chromosome in a single piece. Long read length is critical in providing scaffolding information for de novo genome assemblies. This method also enables the detection of DNA methylation and other medications, essentially for free, with no additional wet lab work. Nanopore sequencing can be used to resolve complex structural rearrangements common with insertions and mobile elements or when verifying constructs. While this technology has a slightly lower per base accuracy than the illumina platform, the ability to generate long sequence reads can offer an advantage for some projects. We offer a full range of data delivery from Promethion, GridION, MinION, MK1C, and flongle devices in house. We have experience creating data for whole genome de novo assemblies for large and small genomes, performing adaptive sampling, CRISPR CAS-9 sequencing enrichment, as well as sequencing plasmids, amplicons, and phages.
Sample requirements: Illumina
DNA quality and quantity for Illumina sequencing
- Many standard kit-based methods for DNA isolation will yield sufficient genomic DNA for Illumina sequencing.
- We recommend the Qiagen genomic isolation kits, Monarch gDNA kits or the Wizard® Genomic DNA Purification Kit.
- We recommend resuspending DNA in 10 mM Tris-HCl pH 8.0 or in molecular-grade water. Please do not resuspend DNA in solutions containing EDTA as EDTA can inhibit downstream reactions. Qiagen EB buffer is okay.
- We recommend that the DNA is quantified using Qubit
- DNA used for Illumina sequencing can be of moderate quality
- DNA used for Illumina sequencing can short fragments above 150bp in size
Submit genomic DNA for illumina sequencing in a clearly labelled (your name and AGXXXX project number) 96-well PCR plate with the DNA purified as follows:
- Please resuspend/elute DNA in water or 10 mM TrisHCl, pH 8.0. Do not use EDTA-containing solutions (e.g., TE buffer) to dissolve or dilute input DNA because EDTA can inhibit enzymatic activity.
- The DNA concentration of each 96 well plate of samples must be measured by PicoGreen, Qubit or other validated dsDNA assay. We strongly advise against using nanodrop
- Minimum Volume: 30 µl DNA to each well of a 96-well PCR plate
- Concentration: each sample must be between 5 ng/µl and 12.5 ng/ul (as measured by QUBIT, not nanodrop), if your samples are outside this range, additional charges will be implemented to normalise samples.
Sample requirements: Nanopore
DNA quality is very important for Nanopore sequencing
DNA and RNA samples from plant, fungal, or avian species are known to contain hard-to-remove and difficult to detect contaminants (e.g. polysaccharides) and/or are inherently difficult to sequence using nanopore sequencing. If you are submitting DNA or RNA from plant, fungal, or avian, please discuss with us first, a pilot project might be advisable using a flongle (mini sequencer) as well as alternative methods for DNA or RNA isolation.
- Spin-column-based methods are generally discouraged as they can result in physically damaged DNA that will produce very short reads and low yields and should not be used for genomic DNA preparation.
- We recommend the Wizard® Genomic DNA Purification Kit (catalog A1120, A1125, A1620 at In Vitro, item PMA1620) or other kits that retain high molecular weight DNA and are not column based.
- We recommend resuspending DNA in 10 mM Tris-HCl pH 8.0 or in molecular-grade water. Please do not resuspend DNA in solutions containing EDTA as EDTA can inhibit downstream reactions.
- We recommend that the DNA is quantified using Qubit
- We recommend the purity of DNA is assessed by Nanodrop (for samples with concentration >20 ng/µl).
- We recommend that sample DNA has a 260/280 around 1.80 and a 260/230 between 2.0-2.2.
Nanopore sequencing devices generate reads that reflect the lengths of the fragments loaded into the flow cell. To have control over the size of the fragments generated in the library prep, it is important to begin with high molecular weight (HMW) DNA.
- A gel image should be submitted of each sample. The gel should contain a size ladder with an upper marker ideally greater than or equal to 20 Kbp (e.g. NEB 1 kb Extended DNA Ladder or Invitrogen 1 Kb DNA Extension Ladder).
The gel should be run slowly and sufficiently long so that it will be possible to determine if sheared DNA exists in the sample.
The shearing of HMW DNA can be minimised by:
- Using wide-bore pipette tips to handle the gDNA. If wide bore pipette tips have not been purchased, it is acceptable to cut the tip off of a regular pipette tip with a new razor blade. The resulting pipette tip opening should be ~3 mm in diameter. All pipetting actions should be performed slowly. Sample mixing by repeated pipetting will shear the sample.
- Using low DNA bind microfuge tubes, such as Eppendorf LoBind tubes.
- Mixing gently but thoroughly by flicking the tube. Do NOT vortex DNA.
- Avoiding unnecessary freeze-thaw cycles
- Keeping the pH between 6 and 9
- Avoiding high temperatures, which can lead to degradation
| Min concentration requirement by Qubit | Min volume | |
| RBK114 kits (Rapid) (over 4 samples) | 20ng/ul | 30ul |
| RBK114 kits (Rapid) (under 4 samples) | 30ng/ul | 30ul |
| LSK114 kits (Ligation) (over 4 samples) | 30ng/ul | 30ul |
| LSK114 kits (Ligation) (under 4 samples) | 85ng/ul | 30ul |
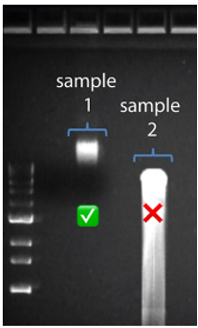
High Molecular Weight DNA (sample 1) shown on the left. Low Molecular weight (Sample 2) on right has significant shearing and is not ideal for nanopore sequencing.
New: Bacterial Genome Assemblies for Nanopore
We offer bacterial genome assemblies
Auckland Genomics offers a custom-built pipeline using the up-to-date methods for bacterial whole genome assembly. See our example bacterial assembly report
Bacterial Genome Assembly example report
https://wujieyun.github.io/bacterial_genome_report/
Briefly, this report provides the following:

- NanoPlot Report
- MultiQC Report
- Data Stats
- Polished Assembly
- Bandage Visualization
- Genome Annotation
- Abricate Output (toxins and AMR genes)
- Assembly Coverage Stats
- Taxonomic Assignment
- Possible Polymorphism HTML Report
- Kraken Report (unmapped reads)
- Krona Plot (unmapped reads)
Not sure which method is right for your project?
Metagenomics is the study of microbial communities in their original habitats and gives a comprehensive insight into the biochemical and metabolic interactions within these communities. Metagenomics can also help identify individual species within microbial habitats with no pre-isolation required. Metagenomics methods are often employed to compare differentially expressed genes within various functional pathways across alternative environments. It reveals the adaptive mechanisms of microorganisms under different environmental stress and explores the interactions between them and other components of their surroundings. There are two main approaches used in metagenomic studies – shotgun-based and amplicon-based. But what exactly are these and how do you decide which to use for your research goals?
16S/18S/ITS Amplicon-based metagenomic sequencing
Amplicon-based metagenomic sequencing efficiently screens for variants and target organisms to describe and compare the diversity of multiple complex environments. The approach is frequently used in population and community microbial ecology studies, phylogenetic reconstruction of target microbial groups, identification of individual species in mixed cultures, and detection of organisms of interest, both pathogenic and beneficial. Amplicon-based metagenomics exploits conserved regions within ribosomal RNA known as amplicons that provide a template for the design of primers to study the variable regions between them. These variable regions are specific to a genus and sometimes a species, meaning that with this method microorganisms can be reliably identified at the genus levels, and some at species levels. The conserved regions used are 16S rRNA, widely used to identify bacteria and archaea; 18S rRNA to identify microbial eukaryotes such as fungi and protists; and ITS sequencing, the preferred method of identification for fungal species.
Shotgun-based metagenomic sequencing
Shotgun-based metagenomic sequencing provides information on the total genomic DNA from all organisms in a sample, avoiding the need for isolation and cultivation of microorganisms or amplification of target regions. This is crucial because it is believed that nearly 99% of all microorganisms cannot be cultivated in the laboratory. Shotgun metagenomic sequencing uses next-generation sequencing to provide information on the genetic diversity of host-associated microbial communities, the functional diversity of microbial communities, gene prediction and annotation, host-microbe interactions, and microbiota-based disease mechanisms.
Metagenomic shotgun sequencing involves randomly shearing the DNA of the microbial genome into small fragments, then adding a universal primer at both ends of the fragments for PCR amplification and sequencing. The sequence of the small fragments is then spliced into a longer sequence through assembly. Because this method sequences the full genome it can provide information to determine not only genus and species but also subspecies and strains in some cases. It can also analyse gene expression and function and how these metabolic functions contribute to community fitness and host-microbe interactions and symbiosis.
Which to use
Which method to use depends on your research goals. Do you want to identify what’s there, or do you want to learn about the functions of what’s there? For example, a large-scale project with the aim of identifying the composition of communities across a range of environments or conditions would likely benefit from amplicon-based sequencing, since it is a much more cost-efficient method than shotgun-based metagenomics and won’t provide metabolic function analysis superfluous to requirement. Amplicon-based sequencing strategies are designed mainly for the purpose of studying the phylogenetic relationship of species, the species composition, and the biodiversity of a microbial community. Besides cost efficiency, other advantages of amplicon-based sequencing include resistance to host DNA contamination, and the risk of false positives is relatively low. The flipside of this is that the resolution is lower and functional profiling is not available.
On the other hand, if your goal is to identify the microorganisms present along with analysing the metabolic functions and dominant pathways within the community, shotgun-based metagenomics would be the approach for you. Apart from the taxonomic analysis that amplicon-based sequencing can provide, shotgun sequencing can also conduct in-depth research on genes and functions of a microbial community, such as pathway analysis using KEGG and GO. This functional profiling comes alongside other advantages such as high resolution, novel gene detection and the estimation of the presence and absence of certain genes and functions. Because of this enriched analysis, shotgun-based sequencing is the more expensive option of the two methods and is also more susceptible to interference from host DNA contamination. It is recommended that host DNA is removed to avoid any extra sequencing costs.
TLDR
In summary, if your goal is to sequence a large number of microbiome samples across different environments or conditions and analyse the diversity of the communities, amplicon-based sequencing is the approach for you.
If you need to analyse the metabolic and biochemical functions within your microbiome samples, shotgun-based sequencing is required.
When starting on a sequencing project, one of the first decisions to make is which sequencing technology to use. Each has its strengths and ideal applications, depending on the specific needs of your research. Here’s a quick guide to help you decide when to choose Nanopore sequencing versus Illumina sequencing.
Nanopore Sequencing: Flexibility and Real-Time Analysis
When to Choose Nanopore Sequencing:
- Long-Read Sequencing: This technology can generate reads that are tens to hundreds of kilobases long, which is particularly beneficial for de novo genome assembly, resolving structural variants, and sequencing repetitive regions that are challenging for short-read technologies.
- Methylation detection. One of the significant benefits of nanopore sequencing is its ability to directly detect DNA methylation or modifcations during the sequencing process. Unlike other sequencing methods, such as bisulfite sequencing, which require DNA to be chemically treated to differentiate methylated from unmethylated cytosines, nanopore sequencing reads native DNA molecules. As a strand of DNA passes through the nanopore, changes in the electrical current are measured, allowing the detection of methylated bases based on the distinct signal changes they produce. This direct approach simplifies the workflow, reduces sample processing time, and minimizes potential biases introduced during chemical conversion. While traditional methods often focus primarily on 5-methylcytosine (5mC), nanopore sequencing can also detect other forms of methylation, such as N6-methyladenine (6mA) and 5-hydroxymethylcytosine (5hmC), in native DNA. This capability allows for a more comprehensive understanding of the epigenetic landscape and its impact on gene expression and cellular function.
- Direct RNA Sequencing: For projects focused on RNA without the need for reverse transcription, nanopore sequencing allows direct RNA sequencing, preserving information like base modifications that could be lost in other methods. However, generally, we recommend Illumina sequencing for standard RNA-sequencing projects.
Illumina Sequencing: Accuracy and Depth
When to Choose Illumina Sequencing:
- High Accuracy and Quality: Illumina sequencing is known for its high accuracy and low error rates, particularly for short-read sequences. If your research requires highly accurate base calling, such as in single nucleotide polymorphism (SNP) detection or other applications where precision is critical, Illumina is the best choice. However, nanopore comes quite close to illumina’s accuracy.
- High Throughput and Scalability: For projects that need large volumes of data, such as whole-genome sequencing of large populations or metagenomics studies, Illumina’s can be more cost efficient. This is typically true when you need hundreds of gigabases or terabases level of data. You can look at our quoting tool yourself to see which method is more cost effective for your project.
- Established Data Analysis Pipelines: If you need access to well-established data analysis pipelines and support, Illumina’s platform has a wealth of tools and resources that can make your data analysis more straightforward and reliable.
Conclusion
Choosing between nanopore and Illumina sequencing depends largely on your specific project needs. If you need long reads, methylation detection, or real-time data, nanopore sequencing is the better option. If high accuracy, high throughput, and cost-effectiveness for large-scale projects are your priorities, Illumina sequencing can be a better choice. Understanding the strengths and limitations of each technology will help ensure you select the best tool for your research objectives.
