
Information about PCR product / amplicon / targeted sequencing services
Amplicon Sequencing
Overview
For this method we use nanopore sequencing to sequence DNA of any length (over 150bp) and we provide the full-length sequence of each DNA molecule.
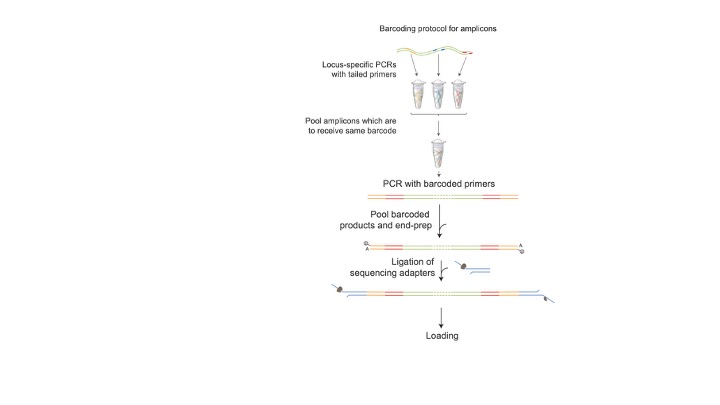
This method works by barcoding each amplicon using PCR. It sequences the full-length sequence read from each molecule. We recommend this method when you are doing 16S metabarcoding or looking at the efficiency of CRISPR edits and want to see the complete target sequence.
For amplicon sequencing using Nanopore, Auckland Genomics can take over at any stage in the process. We can:
- Isolate genomic DNA from samples that meet PC1 requirements.
- We can accept samples that are genomic DNA;
- OR we can accept cleaned PCR products.
Sample requirement for amplicon sequencing:
If you submit DNA samples:
- Concentration: 5ng/ul, measured by Qubit. If your DNA is not at 5ng/ul we will normalise samples for a fee and turnaround times are affected. If DNA normalisation has been included in your quote, please just submit samples at or above 5ng/ul.
- Purity: DNA must be resuspended in molecular grade water or 10 mM Tris-HCl, pH 8.0.
- Volume: at least 20ul total volume, we prefer more.
If you submit amplicons:
- Concentration: amplicons are not over 10ng/ul, and ideally, not under 1ng/ul
- To ensure the most even sequencing coverage across samples, amplicons should be at the same concentration.
- Volume: at least 20ul total volume, we prefer more.
- Purity: amplicons have been purified using a column-based method (PCR clean up kit), bead-based method (AMPure XP beads), or enzymatic method (ExoSap) to remove excess primers and nucleotides.
- Amplicons must have used primers with the following overhangs/tails: Nanopore Forward: TTTCTGTTGGTGCTGATATTGC-[ project-specific forward primer sequence ] 3’
Nanopore Reverse 5’ ACTTGCCTGTCGCTCTATCTTC-[ project-specific reverse primer sequence ] 3’
For full length 16S sequencing using PCR barcoding, use:
ONT_27F: 5’ TTTCTGTTGGTGCTGATATTGC AGAGTTTGATCCTGGCTCAG 3’
ONT_1492R : 5’ ACTTGCCTGTCGCTCTATCTTC TACGGYTACCTTGTTACGACTT 3’
Depending on the species present this will result in a 1400-1600 bp product.
Data delivery:
Varies by kit, usually around ~ >10,000 reads or more per sample
Taxonomic reports:
We also offer taxonomic reports when you sequence the full length 16S rRNA gene. We use the Emu pipeline, original article here: https://www.nature.com/articles/s41592-022-01520-4.
Please see an example report here:
Overview
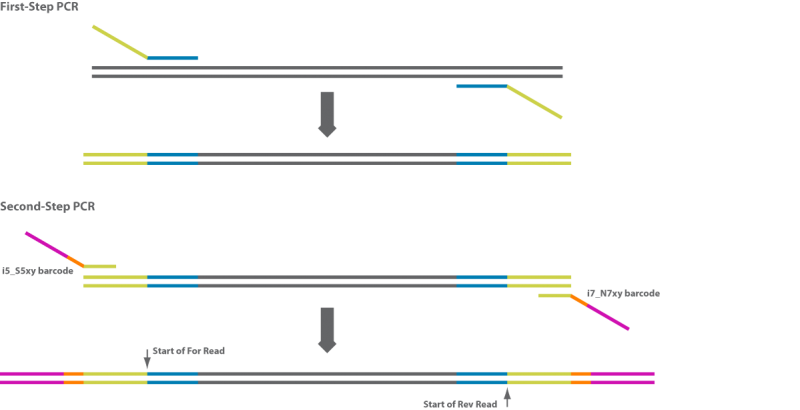
For amplicon sequencing using Illumina, Auckland Genomics can take over at any stage in the process. We can:
- Isolate genomic DNA from samples that meet PC1 requirements;
- We can accept samples that are genomic DNA;
- or we can accept cleaned PCR products.
Sample requirement for amplicon sequencing:
If you submit DNA samples:
- Concentration: 5ng/ul, measured by Qubit. If your DNA is not at 5ng/ul we will normalise samples for a fee and turnaround times are affected. If DNA normalisation has been included in your quote, please just submit samples at or above 5ng/ul.
- Purity: DNA must be resuspended in molecular grade water or 10 mM Tris-HCl, pH 8.0.
- Volume: at least 20ul total volume, we prefer more.
If you submit amplicons:
- Concentration: amplicons are not over 10ng/ul, and ideally, not under 1ng/ul
- To ensure the most even sequencing coverage across samples, amplicons should be at the same concentration.
- Volume: at least 20ul total volume, we prefer more.
- Purity: amplicons have been purified using a column-based method (PCR clean up kit), bead-based method (AMPure XP beads), or enzymatic method (ExoSap) to remove excess primers and nucleotides.
- Amplicons must have used primers with the following overhangs/tails: Forward overhang: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[locusspecific sequence] Reverse overhang: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG‐[locusspecific sequence]
- For 16S sequencing using Illumina, we recommend: Illumina 16S Amplicon PCR Forward Primer = 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG Illumina 16S Amplicon PCR Reverse Primer = 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC, these primers will result in a ~450bp product.
Data delivery:
Varies by kit, usually around ~ >10,000 reads per sample
AmpliconSeq Express Overview:
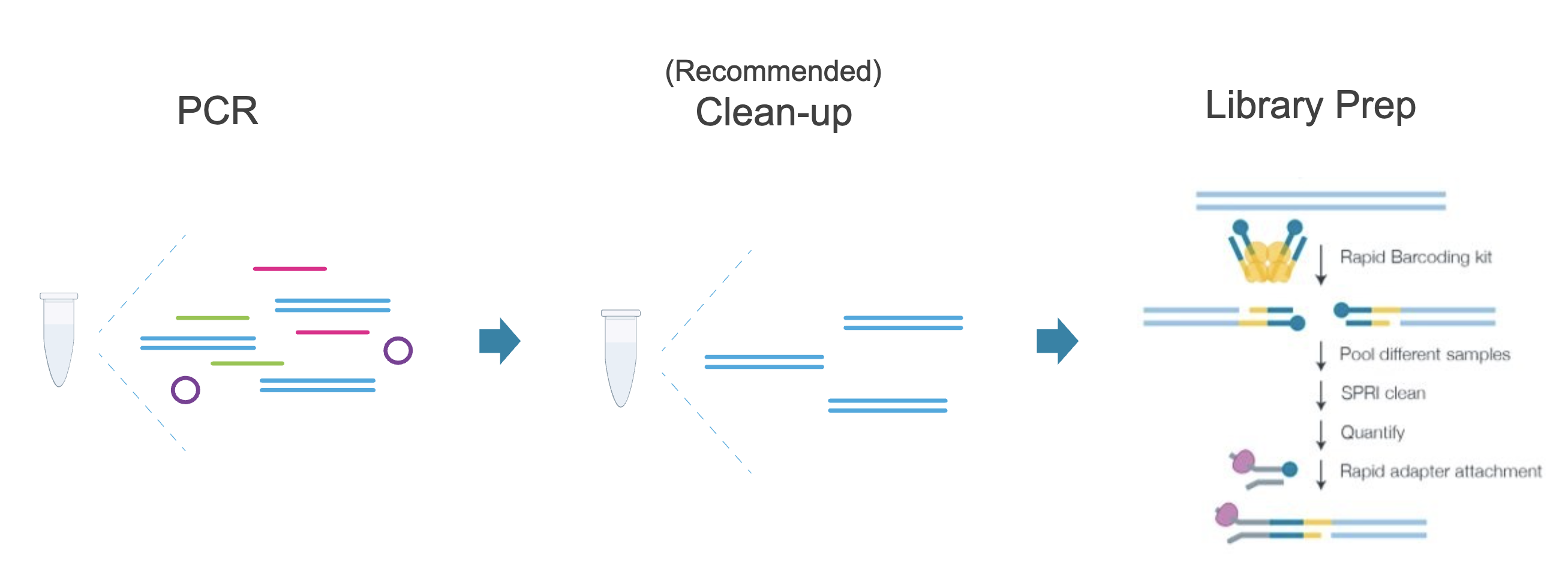
Using Nanopore sequencing, we can also offer a highly cost-efficient approach to sequencing mixed amplicons or multiple PCR products of any length over 150bp. This method randomly cuts each PCR product once and attaches an adapter and barcode at the same time using the RBK114-96 kit from Oxford Nanopore Technologies.
This method works well for PCR products with low diversity or several different low-diversity PCR products pooled together. This approach is commonly used for amplicon sequencing from a single organism (human, mouse, single colony of bacteria, etc). Please get in touch if you need help choosing between the two options (A. and C. here) for amplicon sequencing using nanopore.
Sample Requirements
Submit amplicons for nanopore sequencing DNA in a clearly labelled 96-well PCR plate (your name and AGW-XXX project number) with the DNA purified as follows:
- Concentration: DNA should be at 20ng/ul concentration
- Volume: at least 30ul total volume, we prefer more
- Size: this method works best for amplicons over 200bp in length
- Purity: please clean amplicons up using AMpure beads, PCR clean up kit, ExoSap or similar method before submission, however, we have limited data to indicate un-cleaned PCR products can also work for this method
- Note you do not need to submit primers for this sequencing
Data delivery
On average of 500 reads per sample as default, however, we can generate as much data as you need, please get in touch if you want more data per sample. genomics@auckland.ac.nz
Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. This method sequences a single strand of DNA up to around 800 base pairs in length. All of the DNA in the sample must be the same for this method to work (please no mixed community samples, no 16S from a soil sample).
Genotyping Genotyping is the process of determining the DNA sequence, called a genotype, at positions within the genome of an individual. Sequence variations can be used as markers in linkage and association studies to determine genes relevant to specific traits.
This service can be directly ordered on InfinityX.
Further information around sample requirements and pricing for Sanger sequencing and genotyping can also be found on infinityX or email sequences@auckland.ac.nz if you have questions.
Not sure which targetted sequencing method to use?
Comparison table of options above:
| Feature | Option A: Full-Length Nanopore | Option B: Illumina | Option C: Rapid Nanopore Fragmentation | Sanger Sequencing |
| Read Length | Long read length (up to ~20,000bp) | Short read length (300–600 bp) | Long read length (up to 20,000bp), DNA is cut once randomly | Short (~800 bp) |
| Read Depth | ~ >10,000/sample | ~ >10,000/sample | ~500/sample | one read per sample |
| Primer Requirement | Nanopore overhangs/tails on primers | Illumina overhangs/tails on primers | Standard PCR primers, you do not need to provide us with primers | Standard PCR primers, and you must provide primers to us |
| Best For | 16S full-length, CRISPR, high-diversity | partial 16S, CRISPR, high-diversity | Single gene(s) from one organism, low diversity | Single PCR product per sample, no diversity in sample |
| Accuracy | High | Very high | High | High |
| Throughput | High | High | High | low |
| Ease of Use | Moderate (primer design required) | Moderate (primer design required) | Easy (no custom primers) | Easy (no custom primers) |
| Cost per sample | Medium | Medium | Low | very Low |
| Cost per base | Very low | Very low | Low | High |
Please feel free to get in touch: genomics@auckland.ac.nz if you need any help, we are always happy to chat with you and help you decide what is best for your project.
Not sure whether to use targeted sequencing versus whole genome or metagenomic sequencing?
Metagenomics is the study of microbial communities in their original habitats and gives a comprehensive insight into the biochemical and metabolic interactions within these communities. Metagenomics can also help identify individual species within microbial habitats with no pre-isolation required. Metagenomics methods are often employed to compare differentially expressed genes within various functional pathways across alternative environments. It reveals the adaptive mechanisms of microorganisms under different environmental stress and explores the interactions between them and other components of their surroundings. There are two main approaches used in metagenomic studies – shotgun-based and amplicon-based. But what exactly are these and how do you decide which to use for your research goals?
16S/18S/ITS Amplicon-based metagenomic sequencing
Amplicon-based metagenomic sequencing efficiently screens for variants and target organisms to describe and compare the diversity of multiple complex environments. The approach is frequently used in population and community microbial ecology studies, phylogenetic reconstruction of target microbial groups, identification of individual species in mixed cultures, and detection of organisms of interest, both pathogenic and beneficial. Amplicon-based metagenomics exploits conserved regions within ribosomal RNA known as amplicons that provide a template for the design of primers to study the variable regions between them. These variable regions are specific to a genus and sometimes a species, meaning that with this method microorganisms can be reliably identified at the genus levels, and some at species levels. The conserved regions used are 16S rRNA, widely used to identify bacteria and archaea; 18S rRNA to identify microbial eukaryotes such as fungi and protists; and ITS sequencing, the preferred method of identification for fungal species.
Shotgun-based metagenomic sequencing
Shotgun-based metagenomic sequencing provides information on the total genomic DNA from all organisms in a sample, avoiding the need for isolation and cultivation of microorganisms or amplification of target regions. This is crucial because it is believed that nearly 99% of all microorganisms cannot be cultivated in the laboratory. Shotgun metagenomic sequencing uses next-generation sequencing to provide information on the genetic diversity of host-associated microbial communities, the functional diversity of microbial communities, gene prediction and annotation, host-microbe interactions, and microbiota-based disease mechanisms.
Metagenomic shotgun sequencing involves randomly shearing the DNA of the microbial genome into small fragments, then adding a universal primer at both ends of the fragments for PCR amplification and sequencing. The sequence of the small fragments is then spliced into a longer sequence through assembly. Because this method sequences the full genome it can provide information to determine not only genus and species but also subspecies and strains in some cases. It can also analyse gene expression and function and how these metabolic functions contribute to community fitness and host-microbe interactions and symbiosis.
Which to use
Which method to use depends on your research goals. Do you want to identify what’s there, or do you want to learn about the functions of what’s there? For example, a large-scale project with the aim of identifying the composition of communities across a range of environments or conditions would likely benefit from amplicon-based sequencing, since it is a much more cost-efficient method than shotgun-based metagenomics and won’t provide metabolic function analysis superfluous to requirement. Amplicon-based sequencing strategies are designed mainly for the purpose of studying the phylogenetic relationship of species, the species composition, and the biodiversity of a microbial community. Besides cost efficiency, other advantages of amplicon-based sequencing include resistance to host DNA contamination, and the risk of false positives is relatively low. The flipside of this is that the resolution is lower and functional profiling is not available.
On the other hand, if your goal is to identify the microorganisms present along with analysing the metabolic functions and dominant pathways within the community, shotgun-based metagenomics would be the approach for you. Apart from the taxonomic analysis that amplicon-based sequencing can provide, shotgun sequencing can also conduct in-depth research on genes and functions of a microbial community, such as pathway analysis using KEGG and GO. This functional profiling comes alongside other advantages such as high resolution, novel gene detection and the estimation of the presence and absence of certain genes and functions. Because of this enriched analysis, shotgun-based sequencing is the more expensive option of the two methods and is also more susceptible to interference from host DNA contamination. It is recommended that host DNA is removed to avoid any extra sequencing costs.
TLDR
In summary, if your goal is to sequence a large number of microbiome samples across different environments or conditions and analyse the diversity of the communities, amplicon-based sequencing is the approach for you.
If you need to analyse the metabolic and biochemical functions within your microbiome samples, shotgun-based sequencing is required.
